3.6 KiB
Create
cacmb --create name element_type lattice_parameter esize
Mode create has the following parameters:
name - User defined name that either defines the atom type or the lattice type if using the basis option. If the basis command is not called then name must be an element.
element_type - Specifies which element type to use, this dictates the crystal being build. Current acceptable options for element_type are:
- FCC - Uses the Rhombohedral primitive fcc unit cell as the finite element.
lattice_parameter - The lattice parameter for the crystal structure.
esize - Number of atoms per edge of the finite element. A value of 2 signifies full atomistic resolution and is the lowest number acceptable.
Example
cacmb --create Cu fcc 3.615 11
Creates a copper fcc element with a lattice parameter of 3.615 with 11 atoms per side
Optional keywords
Below are optional keywords that must directly follow after the esize command and before any options or filenames are passed
Orient
orient [hkl] [hkl] [hkl]
The default orientation that is built is [100] [010] [001].
If this keyword is present then the user must provide the orientation matrix in form [hkl] [hkl] [hkl] without spaces.
Currently only accepts whole number values for [hkl].
Example: orient [-112] [110] [-11-1]
Duplicate
duplicate numx numy numz
The default duplicate is 1 1 1.
This is used to replicate elements along each dimension.
The unit of duplication is the lattice periodicity length times the size of elements being duplicated.
numx numy numz dicate the number of duplications units in the x,y, and z dimensions.
This keyword cannot be used with the dim command.
Example: duplicate 10 10 10
Dim
dim dimx dimy dimz
There is no default dim as duplicate is the default option.
This command assigns a box with user-assigned dimensions and fills it with the desired element size.
Note: using dim may not result in periodic cells unless the origin of the cell is shifted from 0,0,0.
Example: dimensions 100 100 100
Origin
origin x y z
This keyword is used to set the origin of the simulation cell.
When using the duplicate command, the origin is set to the minimum position of the rotated element.
When using the dim command the default origin is (0,0,0).
When using the dim command for building it may become necessary to shift the origin of the simulation cell in order to produce a periodic simulation.
This origin can be retrieved by first running --create with duplicate 1 1 1.
Example: origin 10 0 1
Basis
basis basisnum bname bx by bz
This function allows you to define a custom basis to be used at every lattice point. The parameters for this keyword are:
basisnum- The number of atoms in the basisbname- The type of the basis atom (e.g. Cu or Si)bx by bz- The position of the basis atom.
bname bx by bz must be repeated basisnum times for this command.
Example:
basis 2 Si 0 0 0 Si 1.35775 1.35775 1.35775 when used with the fcc element produces a diamond structure with Si.
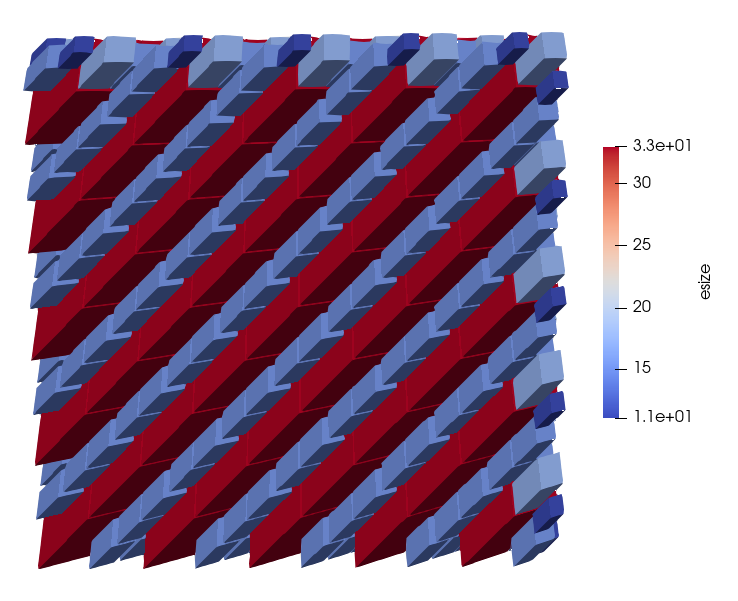
efill
efill
This command attempts to maximize the degree of coarse-graining by iterating through esizes smaller than the user specified when filling in the jagged boundaries to create a periodic block.
This command will iterate through element sizes from the user specified esize-2 to a minimum esize of 11.
Below is an example of a model without efill and a model with efill.
Model without efill

Model with efill